
DNA is the blueprint of life. Genes encode proteins and serve as the body’s basic components. However, building a functioning organism also requires precise instructions about when, where, and how much those components should be produced.
This layer of control is carried out by cis-regulatory elements (CREs), which are short stretches of DNA that serve as binding sites for transcription factors and help control the activity of nearby genes, hence are often described as the “switches” and “dials” of genes.
Although CREs do not encode proteins themselves, they play a major role in shaping traits, guiding development, and influencing disease risk.
CREs control gene expression through epigenetic mechanisms, such as whether DNA is open and accessible and whether it carries markers associated with active gene regulation. Even small changes in CRE sequences can have a substantial effect on gene expression.
A new tool to decode regulation
Until now, scientists have relied on separate experimental methods to study these processes. Some methods identify DNA regions that appear to function as regulatory elements, while others test whether a DNA sequence can activate gene expression. Because these approaches are usually performed independently in different experiments, it has been difficult to directly connect cause and effect or to systematically evaluate the impact of individual changes in the sequence.
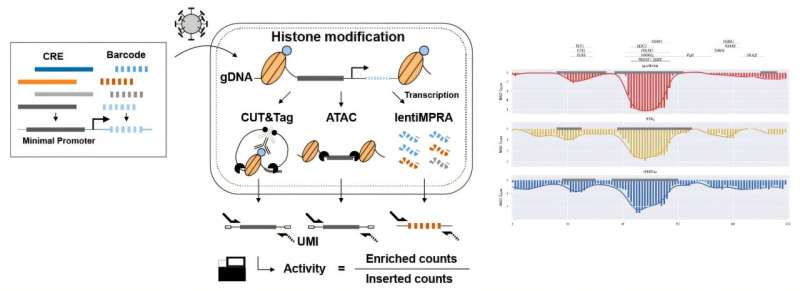
To overcome these limitations, the researchers developed an enrichment followed by epigenomic profiling massively parallel reporter assay (e2MPRA), a new technique that builds on their earlier lentiMPRA platform, which enables simultaneous analysis of thousands of CREs by tagging them with unique DNA barcodes that track their activity.
e2MPRA takes this technique a step further by also capturing epigenetic states, allowing researchers to directly link what a CRE does with how it does it under identical experimental conditions.
Testing thousands of regulatory sequences
e2MPRA was validated using two large libraries totaling approximately 10,000 sequences: one consisted of synthetic CREs with systematically arranged transcription factor binding sites, and the other contained known CREs in which small DNA changes were introduced to examine how each alteration affected function.
For each CRE, the researchers measured three key features: how strongly it activates genes (regulatory activity), whether the surrounding DNA is open and accessible (chromatin accessibility), and whether it carries a chemical “active” mark (H3K27ac modification).
The work is published in the journal Nature Communications.
Distinct regulatory strategies revealed
Using this approach, the team demonstrated that different CREs regulate genes in distinct ways. Some primarily boost gene activity without substantially altering DNA structure, while others mainly increase DNA accessibility. The researchers also found that the arrangement and order of the binding sites within a CRE can strongly influence its activity, much like word order can change the meaning of a sentence.
The team then used e2MPRA to examine how tiny DNA changes (as tiny as a single “letter” difference) can disrupt gene regulation. In regions containing the POU5F1::SOX2 binding site, which plays a key role in maintaining stem cell identity, mutations altered not only gene activity but also DNA accessibility and H3K27ac levels.
In contrast, changes in the YY1 binding site showed a more complex behavior: mutations reduced gene activity but increased DNA accessibility. These findings show that DNA variants can influence gene regulation through multiple, overlapping layers rather than through a simple on-off mechanism.
Implications for variation and disease
“e2MPRA enables us to measure, in parallel and under the same conditions, how mutations in CREs affect both gene activity and epigenetic state, offering a more comprehensive view of gene regulation,” notes Zicong Zhang, first author of the study.
Although the current version of e2MPRA focuses on relatively short DNA sequences and does not yet capture the full three-dimensional organization of the genome, it provides a framework that can be further refined and expanded.
“In 2022, the complete sequence of the human genome was finally decoded. The next challenge is to understand how differences in DNA sequences among individuals lead to differences in gene expression and phenotype,” said Zicong Zhang.
“We expect e2MPRA to become a foundational tool for uncovering the molecular mechanisms behind individual variation and disease risk.”