Highly pathogenic avian influenza A (H5N1, commonly known as HPAI or bird flu) is a formidable worldwide public health concern. With approximately a 100% mortality rate, the World Organization for Animal Health estimates that more than 633 million birds (poultry and wild birds) have been lost globally over the past 20 years. The U.S. Department of Agriculture’s (USDA) Animal and Plant Health Inspection Service (APHIS) estimates that from 2022 (origin date of present outbreak) to April 2025, more than 168 million birds have died, primarily via culling (“depopulation on detection”). Cases are occurring in all 50 U.S. states.
Further, spillovers into other species, such as dairy cows, domestic animals, and even marine mammals, are increasing. For example, by September 15, 2025, bird flu had affected over 1,790 dairy herds across 18 states, according to the Center for Infectious Disease Research and Policy.
Although rare, human spillovers are also occurring primarily from direct exposure to infected animals. A recent report by the U.S. Centers for Disease Control and Prevention (CDC) determined that between March 2024 and May 2025, 70 cases of humans infected with HPAI were documented in the U.S., with one death.
While bird flu primarily devastates avian populations, its occasional spillover into humans raises concern, not only because of the viral infection itself, but also due to the secondary bacterial infections that often complicate severe influenza cases. This intersection of viral and bacterial disease has renewed attention on bacteriophage therapy—not only as a precision tool to combat opportunistic bacterial pathogens, but also as an innovative platform to develop vaccines against HPAI itself.
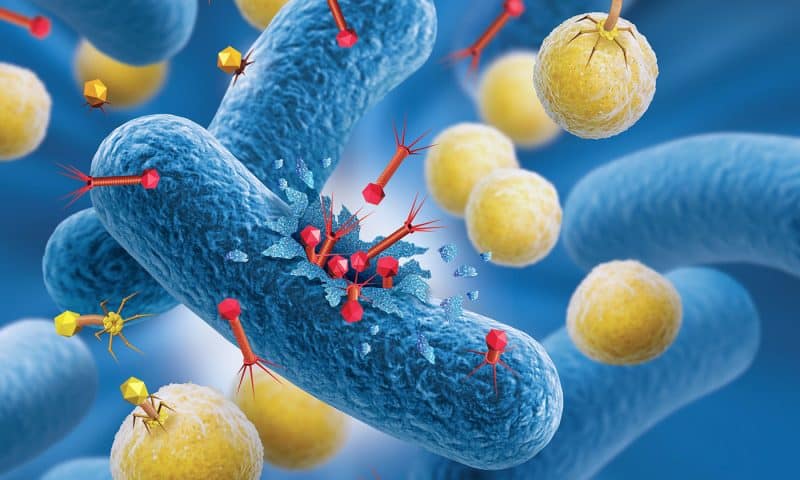
Bacteriophages, or phages for short, are viruses that target and destroy bacteria with remarkable specificity. They consist of genetic material encased in a protein shell that can attach to bacterial cells. Some phages replicate by lysing their hosts, while others integrate into bacterial genomes or persist without lysis, thereby eliciting immune responses from the bacterial host.
In this article, we feature insights from the CDC on the current state of the bird flu epidemic alongside perspectives from companies advancing phage therapeutics as versatile tools for combating resistant bacterial strains and for developing effective, scalable vaccines against viral diseases.
CDC perspective
Gabriel Alvarado, public affairs specialist at the CDC, warns, “Because most people don’t have pre-existing immunity to avian influenza viruses, these viruses have the potential to cause a flu pandemic in people if they were to gain the ability to more easily infect and spread efficiently between people.”
Alvarado says that a recent CDC scientific report made an assessment of the potential pandemic risk of two viruses from human cases and concluded that the risk for this group of viruses is considered moderate. “That assessment validates the value of a continued proactive, coordinated U.S. government response, including continued surveillance and reporting and investigation of every human infection from avian influenza A viruses.”
Phage as vaccine vectors
Amid escalating concerns over avian influenza, the development of vaccine platforms that can be rapidly adapted and scaled has become a priority.
In addition to targeting antimicrobial resistance, Cytophage Technologies also is developing phage-based vaccines against H5N1 using a filamentous phage vector that displays viral epitopes.
“Unlike typical lytic phages, which destroy their bacterial hosts and produce only a short burst of particles, these filamentous phages are extruded continuously,” explains Steven Theriault, PhD, CEO. “The bacteria essentially become miniature phage factories, producing a steady stream of vaccine particles that stimulate the immune system. In this system, we have longevity on our side.”
Theriault believes the key to leveraging phage-based vaccines lies in engineering phage DNA to express antigenic epitopes on the phage surface, with the critical challenge being the selection of stable epitopes. “Viral epitopes can change. To overcome this, we study how the viral shifts occur and carefully select epitopes less likely to be altered by natural evolution.”
According to Theriault, phages as vaccine vectors may offer important advantages. They are self-adjuvanting, strengthening the immune response and reducing or even eliminating the need for booster shots. Manufacturing is also highly efficient—Cytophage estimates it can produce about 150 million doses in seven days. Unlike many existing vaccines, phage-based products can be stored at room temperature, simplifying global distribution. Safety is another key advantage: phages are non-toxic, do not infect human or animal cells, and cannot revert to pathogenic forms.
Theriault says that, unlike other vaccines, phages do not integrate into the host genome, which mitigates risks linked to genomic integration. He also notes that another strength is the speed at which phages can be adapted. “In contrast to the several months required to retool mRNA vaccines, phages can be modified in just a few days. This rapid turnaround could prove decisive in responding to evolving outbreaks like H5N1.”
Dual-action phages
Severe viral infections, such as bird flu, often lead to secondary bacterial infections, particularly in hospitalized or immunocompromised patients. By targeting the specific bacteria responsible, phage therapy could play a crucial role in treating infections, such as bacterial pneumonia. This approach could allow for more tailored and potentially more effective treatments compared to traditional antibiotics, which are often broad-spectrum and face increasing resistance issues.
“Phage-based therapies represent a paradigm shift because, unlike static antibiotics, phages are living biologics that can be thought of as ‘trainable’ antibiotics as they have co-evolved alongside bacteria for billions of years,” notes Amanda Burkardt, CEO, PHIOGEN.
Burkardt says PHIOGEN is developing novel “dual-action” phages that not only fight bacterial infections but also reduce or eliminate recurrences. She reports, “While most phage companies intentionally avoid immune-stimulating phages to reduce the risk of neutralizing antibodies if retreatment is needed, we take the opposite approach; selecting phages that strongly activate the immune system against the infecting pathogen, so patients are, in effect, immunized and protected against future infections without repeated dosing.”
The company’s proprietary technology platform combines directed evolution, high-throughput screening, and immune-relevant models to identify and train phages with enhanced antibacterial and immunogenic properties. Burkardt elaborates, “This is built directly into our technology platform to select for rare phages that both stimulate immunity and remain potent bacterial killers. By systematically harnessing the natural adaptability, we are uncovering entirely new functionalities of phages that antibiotics cannot provide.”
Both of PHIOGEN’s lead candidates emerged directly from this platform. PHI-UI-01, targets recurrent urinary tract infections caused by resistant Escherichia coli, while a parallel program’s candidate, PHI-BI-01, takes aim at extraintestinal pathogenic E. coli (ExPEC). Burkardt discloses, “Our ExPEC bacteremia program is supported by a recent CARB-X (Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator) award and is focused on addressing life-threatening bloodstream infections and delineating the mechanism behind this novel therapeutic and preventative effect. Both programs are based on the same principle of combining immediate bacterial clearance with long-term protection, ensuring a pipeline that addresses both high-prevalence and high-mortality conditions.”
This technology could also help address the pandemic potential of diseases such as bird flu spreading to humans. Burkardt comments, “Our platform is highly relevant to the secondary drug-resistant bacterial infections, like sepsis, that often complicate severe viral illnesses and drive poor outcomes in vulnerable patients. Our dual-action therapeutics offer a two-fold benefit in this setting: (1) immediate treatment of life-threatening bacterial co-infections and (2) reduction in the risk of recurrence or reinfection during prolonged illness or recovery.”
Burkardt concludes, “Our ultimate goal is to build a pipeline of safe, scalable products that not only address today’s resistant and chronic infections but also anticipate tomorrow’s emerging threats.”
Phage cocktails
Armata Pharmaceuticals is developing high-purity, pathogen-specific bacteriophage therapeutics to treat antibiotic-resistant and difficult-to-treat bacterial infections, initially focusing on Staphylococcus aureus (systemic infections) and Pseudomonas aeruginosa (respiratory infections). “Phages are the most ubiquitous organisms on earth,” proffers Sebastien Lemire, PhD, director of Discovery and Engineering. He continues, “Phages are highly species-specific, with a mechanism of action distinct from broad-spectrum antibiotics, that enables phages to bind to and kill specific targeted bacteria while uniquely preserving the normal, healthy human microbiome.”
Armata is developing fixed multi-phage cocktails targeting the desired pathogens. The company utilizes synthetic biology to engineer both phages and manufacturing hosts for improved pharmacological properties. They also employ next-generation sequencing to characterize large proprietary phage libraries.
Lemire elaborates, “Advantages of producing multi-phage cocktails include improved activity against a broad range of clinical isolates and increased genotypic and phenotypic diversity to minimize resistance development.”
Armata has completed three Phase II clinical trials utilizing two distinct phage cocktails, AP-PA02 and AP-SA02, targeting P. aeruginosa and S. aureus, respectively. Deborah Birx, MD, CEO, says that S. aureus plays an especially important role in many secondary post-viral infections. “What we have been able to demonstrate using AP-SA02 in our recent Phase II
diSArm clinical trial is that our phages injected intravenously can home in to the site of infection, penetrate biofilms, infect, and lyse S. aureus. Many deaths post flu, and most likely also potentially from avian flu, are due to bacterial pneumonia and could be treated with phages, as we have shown in complicated bacteremia patients.”
Big data and AI meet wet lab
“Phage therapeutics come with unique challenges,” advises Jonathan Solomon, CEO, BiomX. He continues, “Selecting the right phages requires broad libraries and deep screening, since no single phage works against all clinical bacterial strains. Engineering the phage adds complexity but also opportunity. We need to ensure modifications improve potency, host range, and resistance-avoidance.”
BiomX is addressing those challenges with its BOLT (BacteriOphage Lead to Treatment) platform and in-house expertise. Solomon elaborates, “Our BOLT platform is designed to systematically identify and optimize phage therapies against specific bacterial targets. It works by starting with one of the world’s largest proprietary collections of natural phages, which we screen against thousands of clinical bacterial isolates to identify the most active candidates. We then apply advanced computational and AI tools to predict and evolve phages with improved potency, host range, and the ability to overcome bacterial defense systems.”
Next, the company combines complementary phages into optimized cocktails, balancing lytic activity, biofilm penetration, and resistance prevention to create their targeted therapies. “This integrated approach, where big data and AI meet wet-lab validation, is what enables us to design scalable, highly precise potential phage treatments for some of the most problematic pathogenic bacteria,” says Solomon.
BiomX is advancing its phage therapies in clinical trials against P. aeruginosa and S. aureus. Solomon projects, “What makes these programs especially potentially powerful is that each target represents a ‘pipeline in a product.’ Because these same pathogenic bacteria drive disease across multiple conditions, our validated phage cocktails have the potential to expand into many patient populations.”
And as for bird flu, Solomon says, “Bacteriophages have been at war with bacteria for millions of years, constantly adapting to eradicate them. The bacteria that drive severe secondary infections in bird flu are no exception—there are already phages and phage cocktails with proven activity against these pathogens. In a pandemic-like scenario, where secondary bacterial infections can be deadly, this ability to quickly bring a targeted, validated therapy to market could make phage therapy a uniquely powerful solution.”